Overview¶
dRep is a python program which performs rapid pair-wise comparison of genome sets. One of it’s major purposes is for genome de-replication, but it can do a lot more.
The publication is available at ISME and an open-source pre-print is available on bioRxiv.
Source code is available on GitHub.
Genome comparison¶
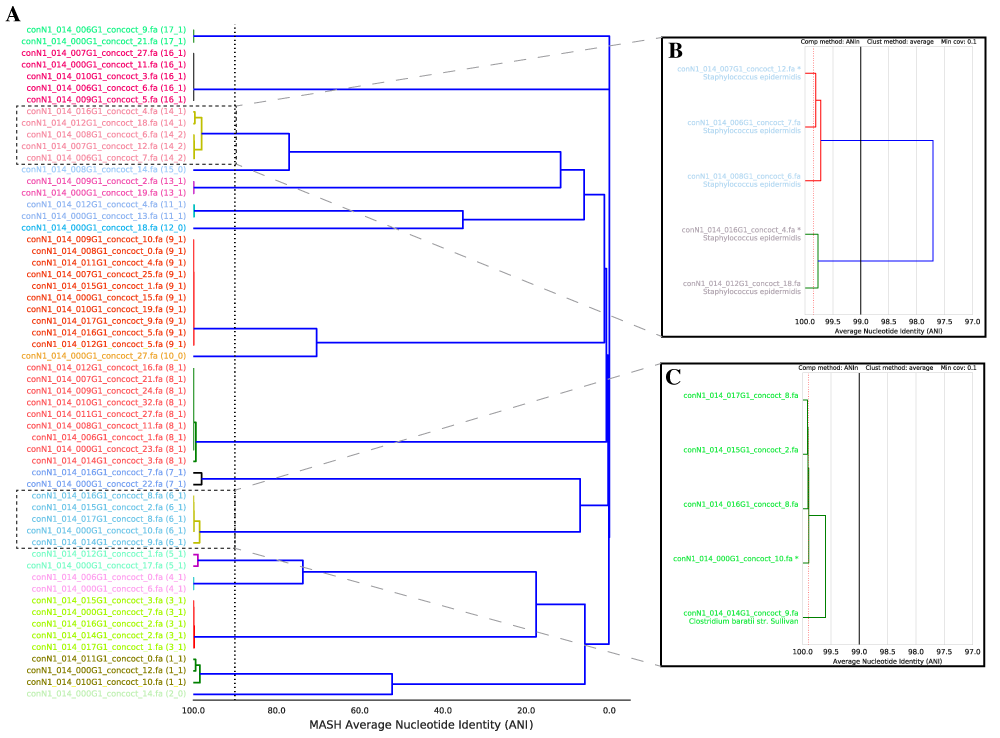
dRep can rapidly and accurately compare a list of genomes in a pair-wise manner. This allows identification of groups of organisms that share similar DNA content in terms of Average Nucleotide Identity (ANI).
dRep performs this in two steps- first with a rapid primary algorithm (Mash), and second with a more sensitive algorithm (ANIm). We can’t just use Mash because, while incredibly fast, it is not robust to genome incompletenss (see Important Concepts) and only provides an “estimate” of ANI. ANIm is robust to genome incompleteness and is more accurate, but too slow to perform pair-wise comparisons of longer genome lists.
dRep first compares all genomes using Mash, and then only runs the secondary algorithm (ANIm or gANI) on sets of genomes that have at least 90% Mash ANI. This results in a great decrease in the number of (slow) secondary comparisons that need to be run while maintaining the sensitivity of ANIm.
Genome de-replication¶
De-replication is the process of identifying sets of genomes that are the “same” in a list of genomes, and removing all but the “best” genome from each redundant set. How similar genomes need to be to be considered “same”, how to determine which genome is “best”, and other important decisions are discussed in Important Concepts
A common use for genome de-replication is the case of individual assembly of metagenomic data. If metagenomic samples are collected in a series, a common way to assemble the short reads is with a “co-assembly”. That is, combining the reads from all samples and assembling them together. The problem with this is assembling similar strains together can severely fragment assemblies, precluding recovery of a good genome bin. An alternative option is to assemble each sample separately, and then “de-replicate” the bins from each assembly to make a final genome set.
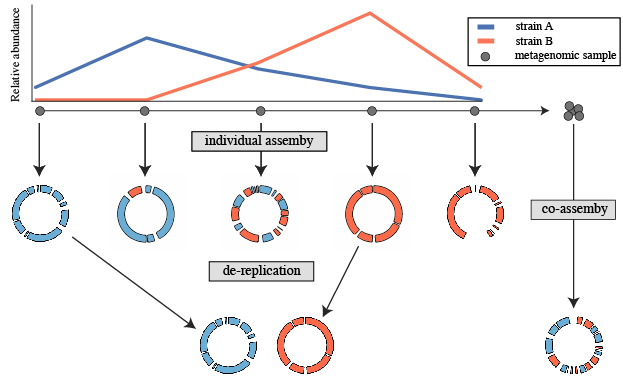
The steps to this process are:
- Assemble each sample separately using your favorite assembler. You can also perform a co-assembly to catch low-abundance microbes
- Bin each assembly (and co-assembly) separately using your favorite binner
- Pull the bins from all assemblies together and run dRep on them
- Perform downstream analysis on the de-replicated genome list
Note
See the publication To Dereplicate or Not To Dereplicate? for an outside perspective on the pro’s and con’s of dereplication
A dRep based metagenomic workflow¶
One method of genome-resolved metagenomic analysis that I am fond of involves the following steps:
- Assemble and bin all metagenomic samples to generate a large database of metagenome-assembled genomes (MAGs)
- Dereplicate all of these genomes into a set of species-level representative genomes (SRGs). This is done with dRep using a 95% ANI threshold.
- Create a mapping database from all SRGs, and map all metagenomes to this database. Each sample will now be represented by a .bam file mapped against the same database.
- Profile all .bam files using inStrain. This provides a huge number of metrics about the species-level composition of each metagenomes.
- Preform strain-level comparisons between metagenomes using inStrain
Some example publications in which this workflow is used are here and there.